




Семинары
Уважаемые коллеги!
На свидетельстве участника семинара, который будет сгенерирован в случае успешного выполнения Вами тестового задания, будет указана календарная дата Вашего он-лайн участия в семинаре.
Семинар "Спинальные мышечные атрофии и боковой амиотрофический склероз, как проявление болезни двигательного нейрона у детей"
Автор: Морозова Т.М.
Проводит: Республиканский Медицинский Университет
Рекомендован по специальностям: Неврология, Педиатрия/Неонатология
Просмотров: 3 188
Дата проведения: с 19.10.2014 по 19.10.2015
Болезнь двигательного нейрона (БДН) охватывает широкий спектр дегенеративных нейромышечных заболеваний, которые сопровождаются гибелью периферических и центральных двигательных нейронов, неуклонным прогрессированием и летальным исходом.
Различные виды БДН отличаются главным образом типом наследования, преимущественной локализацией нейродегенеративного процесса, возрастом начала и скоростью прогрессирования заболевания.
Традиционно к болезням периферического (нижнего) нейрона относят спинальные мышечные атрофии (СМА), а болезням центрального (верхнего) нейрона боковой амиотрофический склероз (БАС).
В МКБ 10 подрубрики СМА и БДН разделены в зависимости от частоты, тяжести заболевания, степени вовлечения различных уровней нервной системы и представлены в рубрике G12 Спинальная мышечная атрофия и родственные синдромы блока «Системные атрофии, поражающие преимущественно центральную нервную систему» (табл.1).
Установление диагноза при этих заболевания ставит много вопросов, как перед родственниками больного, так и перед смежными специалистами. Для комплексного решения этих задач невролог должен обладать современной информацией о течении болезни, патогенезе, классификации фенотипа, прогнозе для больного, генетических ассоциациях, возможности участия в клинических испытания новых препаратов/технических устройств.
- БОЛЕЗНЬ ДВИГАТЕЛЬНОГО НЕЙРОНА – СПИНАЛЬНЫЕ МЫШЕЧНЫЕ АТРОФИИ
Спинальные мышечные атрофии характеризуются дегенерацией периферических двигательных альфа-нейронов спинного мозга и ствола головного мозга. СМА представляют собой гетерогенную группу прогрессирующих заболеваний, генетической природы, которые имеют чаще аутосомно-рецессивный (А-Р), реже аутосомно-доминантный (А-Д) или Х-сцепленный тип наследования. Частота СМА 1 на 6000-10000 новорожденных.
Врачи, выявляющие симптомы слабости и диффузной гипотонии у детей должны иметь в виду высокую вероятность диагноза СМА. Дегенерация мотонейронов в спинном мозге, двигательных ядрах ствола мозга приводит к возникновению симметричного вялого паралича поперечнополосатых мышц конечностей и туловища. В классических вариантах отмечается преимущественное преобладание слабости в проксимальных группах мышц над дистальными, бóльшая слабость в ногах, чем в руках, симметричность снижения силы, отсутствие нарушений чувствительности, снижение или отсутствие сухожильных рефлексов. Как правило, выраженность слабости коррелирует с возрастом начала заболевания.
Наибольшую распространенность имеют проксимальные формы с аутосомно-рецессивным типом наследования, которые составляют 80-85% от всех наследственных СМА. Дистальные спинальные амиотрофии, составляют не менее 10% всех СМА. Учитывая, что распространение мышечной слабости при дистальных СМА сходно с таковым при поражении периферических нервов, ряд авторов обозначает дистальные СМА, как дистальные наследственные моторные нейронопатии (dHMN). Отличительными особенностями дистальных СМА от наследственных моторно-сенсорных нейропатий является: отсутствие чувствительных нарушений, длительная сохранность сухожильных рефлексов, нормальные скорости проведения импульса по периферическим нервам и отсутствие изменения сенсорного потенциала.
Диагностические критерии основных СМА, варианты инфантильных форм СМА, терапевтическую стратегию, модификации, основанные на результатах картирования генов и предродового диагноза, были предложены International SMA Consortium, 2007, 2012 (ISMAC). Наиболее адекватной классификацией является та, в основе которой лежит принцип различия генных мутаций, в том числе в так называемом SMN-гене (ген выживания мотонейрона) (табл.2)..
Таблица 2. Фенотипический спектр проксимальной спинальной мышечной атрофии
|
Тип СМА |
Возраст начала болезни
|
Максимальная функция
|
Естественный возраст наступления смерти |
Типичные проявления
|
|
СМА 0 с артрогрипозом связана с SMN-5q |
врожденная |
Отсутствие движений |
< 30 дней |
Выраженная гипотония, дыхательные нарушения с рождения, диплегия лицевой мускулатуры, наружный офтальмопарез, контрактуры в коленных суставах |
|
Тип I (тяжелая форма) Werdnig-Hoffman связана SMN-5q |
0 – 6 месяцев |
Не сидит |
32% < 2 лет |
Глубокая слабость и диффузная гипотония,нарушение бульбарной функции, слабый крик и кашель, трудность с глотанием и выделением слюны, осложненное течение заболеваний из-за дыхательной недостаточности и аспирационной пневмонии |
|
Тип II (промежу-точная форма) Dubowitz SMN-5q |
7 – 12 месяцев
|
Не стоит |
70% > 2 лет |
Возникновение и прогрессирование слабости в проксимальных отделах конечностей в младенчестве, тремор рук, слабый кашель, дыхательная недостаточность контрактуры суставов и сколиоз. Задержка моторного развития и набора веса, |
|
Тип III а
Тип III б (легкая форма) Kugelberg-Welander связана сSMN-5q |
> 3 года |
Стоит и ходит |
Зрелый возраст |
Мышечная слабость различной степени выраженности, крампи, контрактуры и гипермобильность суставов, повышенный риск переломов, потеря способности ходить с 12 лет в 50% или с некоторого момента жизни |
Для удобства описания проводиться также условное выделение 2 основных групп СМА: изолированные и сочетанные (табл. 3).
|
Таблица 3. Классификация спинальных мышечных атрофий в детском возрасте |
||
|
Нозологические формы |
Тип наследован |
Возраст дебюта |
|
Изолированные формы |
|
|
|
Проксимальные СМА: Аутосомно-рецессивные
СМА 0 -врожденная с артрогрипозом
|
SMN-5q11-q13
SMN-5q11-q13
SMN-5q11-q13 SMN-5q11-q13 |
Врожденная
До 6 мес.
6-12 мес. 1-3года |
|
4.Инфантильная нейрональная дегенерация |
А-Р |
С 1-го мес. |
|
Дистальные СМА: Аутосомно-доминантные Дистальная наследственная моторная нейронопатия тип I Дистальная СМА тип V Дистальная СМА с преимущественным вовлечением верхних конечностей тип V Дистальная СМА нижних конечностей, врожденная, не прогрессирующая Дистальная скапулоперонеальная СМА
Аутосомно-рецессивные Дистальная СМА тип III Дистальная СМА тип IV
Дистальная СМА, X-сцепленная |
7q34-q36 1q13 7p15, 9q34
12q23-q24;
12q24
1p36 11q13 А-Р
Xq13-q21 |
1 -20 1-20 лет Младенческий
Младенческий
Младенческий
Младенческий С рождения до 20 лет С рождения |
|
Сочетанные формы |
|
|
|
Аутосомно-рецессивные Проксимальная СМА с врожденными переломами Проксимальная СМА с пороком сердца Проксимальная СМА с олигофренией и микроцефалией Летальная CMA с контрактурами
Инфантильная СМА с артрогрипозом Бульбоспинальная амиотрофия с глухотой Дистальная инфантильная СМА с диафрагмальным парезом « SMARD » тип VI СМА с понтоцеребеллярной гипоплазией, Норманна синдром Аутосомно-доминантные Скапулоперонеальная СМА с врожденной гипоплазией мышц и параличом голосовых связок тип VII (Harper-Young) Доброкачественная врожденная СМА с контрактурами |
спорадическая А-Р А-Р 9q34, 12q13, 19q13 8q24.3 11q13.2-13.4
14q32 12q23-q24
2q13-14
Хр11.3–q11.2
|
С рождения С рождения С рождения С рождения
С рождения С рождения
1 -2 мес. С рождения
С рождения
С рождения |
Изолированные СМА включают все нозологические формы, при которых симптомокомплекс поражения передних рогов спинного мозга служит ведущим и в большинстве случаев единственным клиническим проявлением. В рамках этой группы традиционно выделяют подгруппы, различающиеся по локализации мышечных атрофий.
Сочетанные СМА подразумеваются те редкие клинические варианты заболеваний, которые характеризуются комбинацией периферического вялого паралича с другой неврологической симптоматикой, а также поражением других органов и систем и необычной комбинацией симптомов.
Электронейромиографический (ЭНМГ) маркер СМА: характерные признаки денервации, вследствие поражения мотонейронов – спонтаннная ритмическая активность (“ритм частокола”), потенциалы фибрилляций, потенциалы фасцикуляций, положительные острые волны, изменение ПДЕ с формированием гигантских полифазных потенциалов, уменьшение числа двигательных единиц.
При морфологическом исследовании мышц выявляются специфические признаки мышечного поражения в виде избыточной неравномерности диаметра мышечных волокон: скопления уменьшенных в размере волокон (“пучковая” атрофия), чередуются с участками гипертрофированных волокон.
ПРОКСИМАЛЬНЫЕ СПИНАЛЬНЫЕ МЫШЕЧНЫЕ АТРОФИИ I ТИПА, II ТИПА И III ТИПА
Заболевание встречается в трех аллельных вариантах, различающихся возрастом начала и тяжестью клинического течения. I вариант описан Werdnig G. в 1891 году, II промежуточный вариант предложил Dubowitz, III вариант – Kugelberg E. и Welander L. в 1956 году. Тип наследования – аутосомно-рецессивный.
Гены, ответственные за возникновение всех трех вариантов заболевания картированы в области хромосомы 5q12.2-q13.3. Эта область представлена инвертированным повтором и включает, по крайней мере, четыре гена, мутации в которых могут иметь значение в развитии заболевания или модифицировать тяжесть его течения. Ген SMN (теломерная копия, MIM: 600354) имеет 9 экзонов, из которых один (8 экзон) не транслируется. Более 95 % больных с I-III вариантами СМА имеют делецию 7 и (или) 8 экзона теломерной копии SMN-гена в гомозиготном состоянии. Остальные пациенты являются компаунд-гетерозиготами и имеют делецию в одном из этих генов, в то время как в другом гене, расположенном на гомологичной хромосоме обнаруживаются сплайсинговые или миссенс-мутации. Мутация в теломерной копии SMN-гена может быть необходимым, но недостаточным условием возникновения заболевания, так как описаны здоровые люди, имеющие такую мутацию в гомозиготном состоянии.
Второй ген, расположенный в этой области – NAIP (ген ингибитора нейронального апоптоза, MIM: 600355) также имеет копии. Данный ген содержит 16 экзонов. Делеции одного или нескольких экзонов этого гена в гомозиготном состоянии встречаются у 40- 70% больных с 1 типом СМА и у 14-22% больных с 2 и 3 типом СМА. У подавляющего числа больных, имеющих делецию 7 и 8 экзонов SMN гена в гомозиготном состоянии, обнаруживается также делеция и в гене NAIP.
Еще один ген, обозначаемый как H4F5 (MIM: 603011), расположен в непосредственной близости от гена SMN, оказывается делетированным у 90% больных со спинальной амиотрофией 1 типа. Предполагается, что этот ген участвует в модификации тяжести клинического течения различных типов проксимальных СМА.
Четвертый ген, вовлеченный в процесс возникновения заболевания – ВTF2p44 также представлен несколькими копиями. Показано, что 15% больных с различными типами СМА имеют делецию этого гена в гетерозиготном состоянии.
Таким образом, ведущим этиологическим фактором проксимальных СМА является наличие делеции в гомозиготном состоянии в теломерной копии SMN гена. Факторами модифицирующими тяжесть течения заболевания и приводящими к возникновению аллельных вариантов проксимальных СМА являются: 1) количество центромерных копий SMN гена (две при I типе СМА и от трех до пяти при II и III типах СМA); 2) наличие делеций в генах NAIP и H4F5. Не исключено существование других модифицирующих механизмов.
Кодируемый геном SMN белок содержит 294 аминокислотных остатка и экспрессируется во всех тканях организма. Наибольшее количество белка обнаружено в мотонейронах спинного мозга. В цитоплазме и ядре соматических клеток SMN ассоциирован с другим белком – SIP1. Вполне вероятно, что этот белок необходим для регенерации и повторных циклов образования мРНК, поскольку при наличии мутации в SMN гене этот процесс у больных нарушен.
I тип проксимальных СМА с аутосомно-рецессивным типом наследования – болезнь Вердниг-Гофманн возникает с рождения до 6-тимесячного возраста и характеризуется тяжелым злокачественным течением. Первые признаки заболевания можно отметить еще во внутриутробном периоде по слабому шевелению плода. При врожденном варианте болезни уже в неонатальном периоде отмечается выраженная мышечная гипотония, гипотрофия с преимущественным поражением проксимальных отделов ног, угасание сухожильных рефлексов, фибриллярные подергивания мышц языка и пальцев кистей. Дети никогда не держат голову и не переворачиваются. Отмечается своеобразная поза ребенка (“поза лягушки”) – конечности отведены в плечевых и тазобедренных суставах и согнуты в локтевых и коленных. Первыми поражаются мышцы проксимальных отделов нижних конечностей, процесс имеет восходящее распространение. Вовлечение дыхательной мускулатуры (межреберных мышц и диафрагмы) приводит к возникновению деформации грудной клетки по типу седловидной, воронкообразной или килевидной, а также сколиоза и кифоза в грудо-поясничном отделе позвоночника. По мере прогрессирования заболевания поражение распространяется на мышцы гортани и глотки вследствие вовлечения в процесс ядер каудальной группы черепных нервов. При этом варианте СМА возможно появление первых признаков болезни в более старшем возрасте, но не позже 6 месяцев. В этих случаях дети могут держать голову и даже переворачиваются, однако, никогда самостоятельно не садятся. Отмечена корреляция между временем появления первых признаков заболевания и продолжительностью жизни. Большинство больных с врожденным вариантом болезни погибают при явлениях сердечной или дыхательной недостаточности, а также от присоединившихся инфекций на первом году жизни. В целом при этой форме СМА продолжительность жизни ограничена 2 годами. Считается, что только 10% – 12% больных переживают пятилетний возраст.
II тип заболевания характеризуется более поздним началом (от 6 до 12 месяцев) и менее злокачественным течением. Для больных характерен период нормального раннего развития: больные удерживают голову, самостоятельно садятся, однако, самостоятельно не ходят. Для этой формы заболевания характерны фасцикулярные подергивания кистей, языка, плечевого и тазового пояса, тремор кончиков пальцев вытянутых рук, контрактуры в суставах и деформации позвоночника. Продолжительность жизни больных увеличена по сравнению с 1 типом болезни и в среднем составляет 10-12 лет.
III тип проксимальных СМA – болезнь Кугельберга-Веландера – возникает в широком возрастном диапазоне от 12 месяцев до 20 лет. Наиболее часто первые признаки болезни появляются в возрасте 2-7 лет в проксимальных группах мышц тазового пояса. Больные начинают испытывать трудности при ходьбе, беге, подъеме по лестнице и подъеме из положения на корточках. В этот период клинические проявления заболевания имеют значительное сходство с таковыми при прогрессирующей мышечной дистрофии Беккера. Сходство дополняется возникновением у 18-25% больных этим вариантом СМА псевдогипертрофийикроножных мышц, а также выраженного лордоза в поясничном отделе позвоночника. Поражение проксимальных отделов рук и плечевого пояса возникают спустя несколько лет после начала заболевания. Также как и для II типа характерно возникновение фасцикулярного тремора кистей, фасцикуляций различных мышечных групп и деформация грудной клетки.
При ЭНМГ- выявляют специфические признаки поражения передних рогов спинного мозга в виде формирования “ритма частокола” – спонтанной ритмической активности.
При морфологическом исследовании биоптатов мышечных волокон выявляются атрофированные и гипертрофированные волокна 1 и 2 типа. Характерным признаком является скопление мелких круглых волокон, чередующиеся с гипертрофированными волокнами (“пучковая” атрофия). При патоморфологическом изучении выявляется набухание, сморщивание или атрофия мотонейронов передних рогов спинного мозга, а в ряде случаев ядер черепных нервов.
Проводится дородовая диагностика с использованием методов ДНК-анализа, направленных на обнаружение делеций 7 и 8 экзонов SMN-гена в гомозиготном состоянии. Возможно обнаружение гетерозиготного носительства делеции в гене SMN.
Спинальная амиотрофия Х-сцепленная летальная
Впервые описана Baumbach и соавт., в 1994 году. Учитывая, что это вторая форма спинальных амиотрофий с Х- сцепленным рецессивным типом наследования, описанная после спино- бульбарной амиоторофии Кеннеди, эту форму иногда обозначают как СМА Х2. Тип наследования Хр-сцепленный рецессивный.
Клинические проявления возникают с рождения и характеризуются симптомами вялого паралича, с преимущественным поражением мышц плечевого и тазового пояса и проксимальных отделов конечностей в сочетании с врожденными, или появляющимися в период новорожденности, контрактурами крупных суставов. В целом клинические проявления заболевания, а также морфологические и ЭНМГ признаки полностью соответствуют таковым при проксимальной спинальной амиотрофии Верднига-Гоффмана.
Врожденная цервикальная спинальная мышечная атрофия.
Редкая форма врожденной патологии впервые описана в 1981 году. Заболевание обнаруживается сразу после рождения и проявляется в виде атрофии мышц рук, мышечной гипотонии, ограничения двигательной активности и отсутствия сухожильных рефлексов. Мышцы нижних конечностей не страдают.
Инфантильная нейрональная дегенерация.
Это заболевание принято описывать как инфантильную спинальную мышечную атрофию, сочетающуюся с дегенеративными изменениями в мозжечке и таламусе. В клинической картине не первый план выступает гипотония и арефлексия, диспноэ, слабый крик, затрудненной сосание и глотание. Если симптомы болезни обнаруживаются с рождения, смерть наступает в пределах 5 мес.
Инфантильная СМА с артрогрипозом (врожденный нейрогенный артрогрипоз).
Термин нейрогенный артрогрипоз подразумевает сочетание артрогрипоза с инфантильной спинальной мышечной атрофией, обусловленной первичной дегенерацией клеток передних рогов спинного мозга, передних корешков и/или периферических нервов. Большинство описанных случаев спорадические. В клинической картине у новорожденных общая гипотония, отсутствие движений, респираторные расстройства, трудности сосания и глотания (аспирация), фациальная диплегия, наружная офтальмоплегия, деформация суставов рук и ног, ограничивающая движения. Выражен респираторный дистресс с парезом диафрагмы. Сухожильные рефлексы снижены или отсутствуют. Переломы с рождения, фациальные дизморфии. Продолжительность жизни менее 2 лет. Рентгенологически – релаксация купола диафрагмы.
Таблица 4. Особенности Х-сцепленной СMA в сравнении с SMN1 связанные СMA и непрогрессирующим артрогрипозом
|
Особенности |
XL-СMA |
Аутосомно-рецессивный тип СMA |
Артрогрипоз |
||||
|
0 |
I |
II |
III |
IV |
|||
|
Множественные контрактуры |
– |
– |
– |
– |
|||
|
Переломы |
± |
± |
– |
– |
– |
– |
± |
|
Гипотония |
– |
± |
|||||
|
Мышечная слабость |
± |
||||||
|
Моторная регрессия |
± |
± |
– |
– |
|||
|
Нормальные когнитивные функции |
|||||||
|
Отсутствие сухожильных рефлексов |
(70%) |
± |
± |
± |
|||
|
Миопатическое лицо |
± |
± |
– |
– |
– |
– |
– |
|
Нейрогенная атрофия |
± |
||||||
|
Денервационный тип ЭНМГ |
± |
||||||
|
Утрата клеток переднего рога |
± |
||||||
Спинальная амиотрофия Рюкю
Впервые описали Кondo и соавт., в 1970 году у 32 больных из высокоинбредной популяции на островах Рюкю в Японском море.
Тип наследования аутосомно-рецессивный. Заболевание не сцеплено ни с одним из известных генов, приводящих к возникновению спинальных амиотрофий. Первые признаки заболевания появляются вскоре после рождения и характеризуются симметричной слабостью мышц проксимальных отделов ног и фасцикуляциями. Отсутствие рефлексов. Ноги поражаются больше рук. По мере прогрессирования заболевания отмечается распространение патологического процесса на мышцы дистальных отделов ног, с формированием полой стопы. Дети сидят, стоят и иногда ходят
Мышцы проксимальных отделов рук поражаются спустя несколько месяцев. У 8 больных выявлены фибрилляции мышц. В большинстве случаев у больных обнаруживается микроцефалия, кифосколиоз и частичная кожная синдактилия на кистях и стопах. Частые респираторные заболевания. Микрокрания, но когнитивных нарушений нет.
На ЭМГ выявляются типичные признаки поражения мотонейронов спинного мозга.
СМА с понтоцеребеллярной гипоплазией; А-Р; болезнь Нормана
Клинические признаки: Дебют пренатально или с рождения, слабое шевеление плода, гипотония, мышечная слабость, снижение спонтанной двигательной активности, арефлексия, психомоторная ретардация, атаксия, нистагм. Могут быть контрактуры, иногда признаки артрогрипоза. Трудности вскармливания. Различают 2 типа болезни Нормана:
тип 1: – глобальная церебеллярная гипоплазия смерть к 1 году
тип;
2: – прогрессирующая микроцефалия, экстрапирамидная дискинезия, эпилепсия, ментальная ретардация.
МРТ: мозжечковая и понтинная гипотрофия, кортикальная атрофия
Проксимальная СМА I типа с врожденными переломами
Клинические признаки: гипотония. Респираторные трудности. Гипотрофия мышц. Костные аномалии: Генерализованная остеопения, врожденные переломы. Множественные контрактуры. Врожденный порок сердца. Гипертрихоз. Смерть в младенчестве.
ОБЩАЯ ХАРАКТЕРИСТИКА ДИСТАЛЬНОЙ СМА.
Первые симптомы появляются в 6 – 24 мес. в виде билатерального пареза ног. Дистальная слабость перонеальных мышц и разгибателей пальцев ног, гипотония и атрофии, снижение и/или отсутствие сухожильных рефлексов. Деформации стоп. Трудности при ходьбе и частые падения. Заболевание медленно прогрессирует с распространением на проксимальные отделы конечностей ног и рук, мышцы туловища и диафрагмы. Однако, всегда слабость превалирует в ногах и дистальных отделах конечностей. Дыхательные нарушения прогрессируют до острого респираторного дистресса. Парез мышц туловища ведет к гиперлордозу, сколиозу. Деформации грудной клетки нет. Больные имеют нормальный интеллект. Краниальные мышцы интактны, чувствительных нарушений, фасцикуляций, пирамидных знаков нет.
Спинальная амиотрофия врожденная непрогрессирующая с преимущественным поражением ног
Впервые описана Frijns et al. в 1994 году. Тип наследования аутосомно-доминантный. Ген локализован в хромосоме 12 q23-q24.
Первые проявления заболевания обычно выявляют к 15-18 месяцам жизни по задержке темпов приобретения навыков самостоятельной ходьбы. Обычно дети начинают самостоятельно ходить после полутора лет и имеют варусную деформацию стоп. Походка больных напоминает степпаж. По мере прогрессирования заболевания формируется эквиноварусная установка стоп и отмечается восходящий тип распространения мышечной слабости, с постепенным вовлечением отводящих мышц бедер и формированием контрактур в коленных суставах. Сухожильные рефлексы не изменены. Чувствительных нарушений не выявляется. Ни у одного больного не отмечено слабости мышц рук и туловища. У ряда больных спустя 15-20 лет от начала заболевания может появляется умеренно выраженная слабость лицевой мускулатуры и сгибателей шеи.
Характерным ЭНМГ признаком заболевания являются гигантские моторные потенциалы действия при нормальных значениях скоростей проведения импульса по моторным и сенсорным волокнам периферических нервов.
У некоторых больных обнаруживаются пограничные с нормой значения активности креатинфосфокиназы. При морфологическом исследовании биоптатов мышц выявляется характерная для спинальных амиотрофий “пучковая” атрофия волокон, преимущественно 1 типа.
Спинальная амиотрофия с глазодвигательными нарушениями и эпилепсией.
Заболевание описано Oка с соавт. в 1995 году. Тип наследования аутосомно-рецессивный
Первые проявления отмечаются в период новорожденности и характеризуются прогрессирующей генерализованной спинальной амиотрофией, с характерными фасцикуляциями скелетных мышц и языка, сухожильной гипорефлексией и гипотонией. У детей часто возникают колебания температуры тела. К 6 годам к указанным симптомам присоединяются глазодвигательные нарушения в виде наружной офтальмоплегии и тонико-клонические судороги. При проведении магнитно-резонансного исследования мозга обнаруживается гипоплазия мозжечка и генерализованная атрофия коры головного мозга. Интеллект больных снижен.
На ЭМГ выявляются типичные изменения мотонейронов передних рогов спинного мозга.
Морфология. Избыточная неравномерность диаметра мышечных волокон.
Врожденная скапулоперонеальная спинальная амиотрофия
Впервые описал De Long & Siddique в 1992 году. Тип наследования аутосомно-доминантный. Ген заболевания, обозначаемый как SPSMA (скапуло-перонеальная спинальная мышечная атрофия) картирован на хромосоме 12q24.1-24.31.
Заболевание возникает с рождения и характеризуется врожденной гипоплазией мышц, симптомами спинальной амиотрофии, наиболее выраженных в области надостных и подостных мышц и мышц перонеальной группы. У большинства больных возникает стридорозное дыхание, обусловленное параличом голосовых связок. По мере прогрессирования заболевания признаки мышечной слабости и атрофии распространяются на мышцы дистальных отделов рук и ног. Во 2 или 3 декаде жизни возникают контрактуры в крупных суставах, чаще всего нижних конечностей. Течение заболевания умеренно прогрессирующее. У мужчин заболевание течет тяжелее, чем у женщин. В сегрегирующих семьях наблюдается антиципация.
На ЭМГ выявляются типичные изменения мотонейронов мотонейронов передних рогов спинного мозга.
Морфология: Избыточная неравномерность диаметра мышечных волокон.
Дородовая диагностика возможна в сегрегирующих семьях на основании изучения сцепления с ДНК маркерами области хромосомы 12q24.
Инфантильная спинальная амиотрофия с параличом диафрагмы – SMARD
Впервые описали Mellins и соавт., в 1976 году. Тип наследования аутосомно-рецессивный. Ген заболевания является геном иммуноглобулин-связывающего белка (IGHMBP2, MIM: 600502) и картирован на хромосоме 11q13-q13.4. Основное количество мутаций найдено в экзоне 5 (миссенс, нонсенс, сдвиг рамки считывания).
Заболевание составляет 1% всех случаев инфантильных спинальных амиотрофий. Первые проявления заболевания возникают с рождения или в периоде новорожденности и характеризуются симптомами вялого паралича или пареза, наиболее выраженными в мышцах дистальных отделов верхних конечностей. В ряде случаев первые признаки заболевания отмечаются еще во внутриутробном периоде и характеризуются снижением двигательной активности плода. Отличительными особенностями этой формы болезни является паралич и эвентрация диафрагмы, приводящие к возникновению выраженных дыхательных расстройств, а также фасцикуляции языка.
Дыхательные нарушения могут быть столь значительными, что требуют искусственной вентиляции легких и являются основной причиной гибели больных в возрасте до 3 месяцев. У большинства больных уже в период новорожденности формируются контрактуры в коленных и голеностопных суставах.
На ЭМГ выявляются признаки поражения клеток переднего рога спинного мозга.
Истощение и гибель мотонейронов спинного мозга. В биоптате мышечных волокон выявляется феномен «пучковой» атрофии мышечных волокон без признаков реиннервации. Возможна дородовая диагностика.
Бульбоспинальная амиотрофия с глухотой
(болезнь Brown-Vialetto-Van Laere)
У детей представлен аутосомно-рецессивный вариант SLC52A2; 8q24 с нарушением транспорта рибофлавина. Первые симптомы появляются в 2-5 лет. Выявляют билатеральную сенсоневральную глухоту, атрофию зрительного нерва, клинику поражения 7, 9-12 пар черепных нервов (бульбарный синдром), фасцикуляции языка, гиповентиляцию. С течением времени паралич распространяется на плечевой пояс, руки, грудную клетку. Возникает диафрагмальная слабость, отсутствуют рефлексы. Формируется сколиоз. ЭНМГ: денервация с элементами аксональной полиневропатии. Снижен уровень карнитина. В терапию необходимо включать рибофлавин 100-500 мг/день
Бульбарный парез Фацио-Лонде
Эта форма представлена аутосомно-доминантным и аутосомно-рецессивным вариантами SLC52A3; 20p13. Заболевание возникает в возрасте от 1 года до 12 лет и характеризуется симптомами бульбарного паралича или пареза, слабостью лицевой мускулатуры, птозом и нарушением подвижности диафрагмы. При аутосомно-рецессивном варианте заболевания отмечаются прогрессирующий стридор, выраженные дыхательные расстройства, значительное слюнотечение, генерализованная гиперрефлексия, паралич диафрагмы. Этот вариант характеризуется более ранним началом и злокачественным течением, приводящим к смерти больного в течении двух лет от начала заболевания. При аутосомно-доминантном типе наследования заболевание возникает позднее и характеризуется нерезко выраженными дизартрией и дисфагией, умеренным птозом, слабостью мышц лица, отсутствием дыхательных нарушений и медленным прогрессированием.
На ЭМГ выявляются признаки денервации и увеличение амплитуды М- ответа. При проведении патоморфологического исследования выявляются дегенерация и истощение нейронов черепно-мозговых нервов, спинного мозга и мозжечка.
ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ ИНФАНТИЛЬНЫХ И ДЕТСКИХ ФОРМ СМА
Дефицит кислой мальтазы (гликогеноз II типа)
Адренолейкодистрофия
Ботулизм
Наследственные мото-сенсорные невропатии
Синдром Дауна
GM1 англиозидоз
Синдром Хурлера
Болезнь Гоше
Синдромы Марфана и Прадера Вили
Метаболические болезни (органические ацидурии и митохондриальные болезни)
Неонатальная и врожденная миастения
Периферические невропатии
Гликогеноз( болезнь Помпе)
Полиомиелит
Поражение спинного мозга
Лечение СМА предполагает средства направленные на сохранение мотонейрона (рилузол, вальпроевая кислота), улучшение энергетической обеспеченности (L-карнитин, коэнзим Q10), использование клеточной терапии.
Алгоритм диагностики СМА представлен на рис. 1.
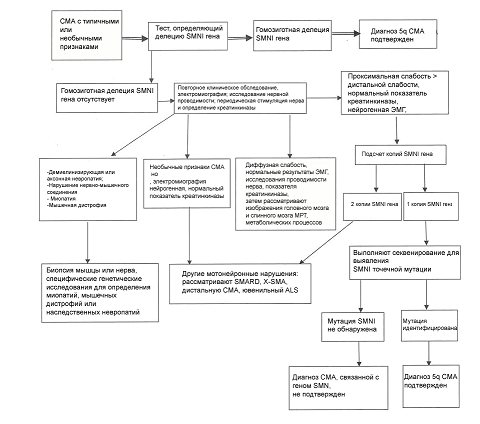
{kind=link}
Рисунок 1. Алгоритм диагностики СМА (по Consensus Statement for Standard of Care in Spinal Muscular Atrophy, 2007
- БОЛЕЗНЬ ДВИГАТЕЛЬНОГО НЕЙРОНА У ДЕТЕЙ – БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
В последнее время отмечены тенденции к росту заболеваемости БДН во всех возрастных группах. Распространенность БДН в мире в среднем составляет 2–5/100 тыс. человек в год. Согласно классификации F.Norris в 80% случаев БДН представлена боковым амиотрофическим склерозом (БАС), в 10% –прогрессирующим бульбарным параличом, а также двумя редкими формами: в 8% – прогрессирующей мышечной атрофией (изолированное медленно прогрессирующее поражение ПМН) и в 2% – первичным боковым склерозом (изолированное медленно прогрессирующее поражение центрального двигательного нейрона). К клиническим проявлениям БДН относят признаки поражения периферических мотонейронов (ПМН), такие как парезы и атрофии скелетных мышц с фасцикуляциями в них, а также признаки поражения центральных мотонейронов (ЦМН), такие как спастичность, гиперрефлексия, патологические пирамидные знаки при длительной сохранности брюшных рефлексов (за исключением определенных фенотипов болезни). БДН в дебюте заболевания или по мере ее прогрессирования сопутствуют бульбарный и псевдобульбарный синдромы, признаками которых являются вялая или спастическая дизартрия, дисфагия, атрофия языка и фасцикуляции в нем, возможное повышение нижнечелюстного и глоточного рефлексов, ларингоспазм и насильственный смех и плач. Распределение и выраженность симптоматики поражения ПМН и ЦМН в значительной степени зависят от формы, дебюта и варианта болезни. На завершающей стадии болезни у пациентов развиваются стволовые или спинальные дыхательные нарушения, которые наряду с дисфагией и алиментарной недостаточностью являются причиной летального исхода
У детей БДН ассоциируется с генетическими мутациями, представленными в таблице 5.
Диагноз БДН необходимо подтвердить инструментально с помощью ЭНМГ и МР-визуализации. Задачей этих методов является исключение других заболеваний центральной и периферической нервной системы, которые потенциально излечимы и имеют доброкачественный прогноз.
ЭНМГ: при игольчатой миографии на трех уровнях (голова или шея, рука, нога) в наиболее пораженных мышцах выявляется спонтанная
активность в виде потенциалов фасцикуляций, фибрилляций и положительных острых волн, а также тенденция к увеличению длительности, амплитуды и количества фаз потенциалов двигательных единиц (признаки нейрональной денервации). В начальных стадиях болезни спонтанная активность с преобладанием фасцикуляций сочетается со снижением длительности потенциалов двигательных единиц. На начальных стадиях при глобальной миографии в покое, тонических пробах и расслаблении в мышцах больных регистрируются потенциалы фасцикуляций с частотой 1–2 Гц при нормальном интерференционном паттерне кривой максимального усилия.
В развернутой стадии болезни в покое, тонических пробах и расслаблении отмечаются ритмичные высокоамплитудные потенциалы фасцикуляций, а при максимальном усилии – ритм “частокола”. Выбирая мышцы для исследования методом игольчатой ЭНМГ, следует помнить, что раньше всего при БДН страдают мышцы разгибательной группы (группа локтевого и лучевого нервов на руке и малоберцового нерва на ноге). При стимуляционной ЭНМГ на трех уровнях отмечается снижение амплитуд М-ответов, уменьшение скоростей проведения по двигательным волокнам периферических нервов, но не более чем на 30%, сохранность потенциалов действия нервов и скоростей проведения по чувствительным волокнам (критерии ЭНМГ-диагностики БДН Ламберта) и увеличение соотношения амплитуд Н-рефлекса и М-ответа в икроножных мышцах, что отражает наличие пирамидного синдрома
Больным необходимо проводить МРТ хотя бы двух отделов центральной нервной системы – ЦНС (на уровне, пораженном в дебюте заболевания, и уровне, наиболее близком к дебюту заболевания).
ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ БАС:
СМА
Болезнь Тея-Сакса
Миастения
Плексопатия
ОМАН
Полиомиелитические синдромы
Синдром Хопкинса
Паранеопластические синдромы
Токсическое влияние (свинец)
Терапия БАС является посиндромной: защита мотонейрона (рилузол), уменьшение спастичности (баклофен, мемантин), слюнотечение (имипрамин), улучшение метаболизма (L-карнитин, коэнзим-Q10).
Курирующий врач, также, должен составить вместе с семьёй план многопрофильного наблюдения ребенка. Этот план обычно включает консультирование и оказание помощи у пульмонолога; гастроэнтеролога/диетолога; ортопеда/реабилитолога. А также, детского невролога (если ребенок наблюдается педиатром или семейным врачом, а не неврологом); медицинского генетика (при необходимости решения вопросов планирования семьи).
Пациенты с БМН с течением времени постепенно теряют мышечную силу и подвижность. Комплексная реабилитация поможет задержать или предотвратить вторичные нарушения, включая мышечные контрактуры, замедлит прогрессирование заболевания, а также сохранить или потенциально улучшит мышечную силу и двигательные функции. В реабилитационные мероприятия, при СМА и БДН включают физиотерапию, трудотерапию, логопедию, использование ортопедического и другого адаптивного оборудования, общеукрепляющее упражнения и упражнения на равновесие, растяжение или увеличивающие диапазон движений, водной терапии, физической и социальной активности. Реабилитационные мероприятия, должны обеспечить дополнительную помощь, направленную на улучшение дыхательной функции и питания.
Таблица 5. Фенотипические варианты БАС у детей
|
Форма |
Белок |
Насле-дование |
Клинические варианты, дебют |
Клинические признаки |
|
БАС 2 |
Alsin |
А-Р; 2q33 |
Ювенильный первичный боковой склероз После 10 лет |
Прогрессирующие признаки поражения ЦМН снижение возможности к передвижению во второй декаде жизни, нарушение моторной речи и когнитивных функций |
|
Ювенильный амиотрофический боковой склероз После 6 лет |
Спастика лицевой мускулатуры, псевдобульбарный синдром, спастическая параплегия. Дистальные амиотрофии – в ногах больше, чем в руках. Медленно прогрессирует. Редукция ходьба после 40 лет. |
|||
|
Spatacsin; |
А-Р; 15q21 |
7-23 года |
Спастичность, псевдобульбарный синдром, гиперрефлексия. Дистальные амиотрофии кистей и стоп, атрофия языка, фасцикуляции, слабость и атрофии предшествуют спастичности. Медленно прогрессирует. |
|
|
|
|
А-Р; 6p25, 21q22 |
4-10 лет |
Умеренный птоз. Дистальная симметричная атрофия кистей и стоп. Бульбарный синдром, слабость лицевых мышц, дисфазия. Спастичность рук и ног. Снижение слуха. Гинекомастия. |
|
БАС 16 |
Sigma-1 Receptor |
А – Р, 9p13.3 |
1-2 года |
Спастичность ног больше рук Слабость в кистях. Утрата передвижения к 3-й декаде жизни. |
|
БАС 4 |
Senataxin |
А-Д,9q34.13 |
6-21 год |
Раннее нарушение походки. Слабость дистальная, позже проксимальная. Бульбарные нарушения редко. Гиперрефлексия 78%, атрофия азыка 17%. Медленно прогрессирует. |