



Семинары
Уважаемые коллеги!
На свидетельстве участника семинара, который будет сгенерирован в случае успешного выполнения Вами тестового задания, будет указана календарная дата Вашего он-лайн участия в семинаре.
Семинар "МИОТОНИЧЕСКОЕ РАССТРОЙСТВО: ВРОЖДЕННЫЕ МИОТОНИИ И МИОТОНИЧЕСКАЯ ДИСТРОФИЯ"
Автор: Морозова Т.М., Морозова А.В.
Проводит: Республиканский Медицинский Университет
Рекомендован по специальностям: Неврология, Семейная медицина/Терапия
Просмотров: 6 407
Дата проведения: с 24.12.2014 по 24.12.2015
Врожденные миотонии и миотоническая дистрофия относятся к миотоническим расстройствам (МКБ Х – G71.1).
Общим клиническим симптомом миотонических расстройств является миотонический феномен, характеризующийся возникновением после произвольного сокращения тонического спазма мышц с задержкой последующей миорелаксации. Фаза расслабления мышц затягивается на 5–30 сек. Миотонический спазм особенно провоцирует быстрое движение, совершаемое со значительным усилием, например трудность в освобождении руки после рукопожатия или невозможность быстро открыть глаза после зажмуривания.
В широком смысле под миотоническим синдромом понимают любое нарушенное расслабление мышц с мышечным спазмом, которое не относится к пирамидно-экстрапирамидной ригидности, и может иметь как транзиторный, так и перманентный характер.
При классических формах миотонического расстройства миотонический феномен возникает в начале активных движений и уменьшается или исчезает при последующих мышечных сокращениях «феномен разминки».
Для большинства нозологических форм миотонии характерны истинные мышечные гипертрофии. Они наблюдаются при миотонии Томсена, Беккера, перманентной миотонии, флюктуирующей миотонии.
Ряд форм наследственных миотоний сопровождается симптомами прогрессирующей мышечной слабости и гипотрофии. Типичным примером таких заболеваний является миотоническая дистрофия Куршмана-Штейнерта-Баттена и проксимальная миотоническая миопатия Рикера.
Помимо классических вариантов выделяют так называемые парамиотонии или парадоксальные миотонии, при которых миотонический феномен появляется после периода активных мышечных сокращений. В большинстве случаев возникновение парамиотоний вызывается холодом, однако выявлен ряд форм, интактных к холодовым воздействиям.
Отдельно выделяют эпизодические миотонии (флюктуирующая миотония, ацетазолзависимая миотония, волнообразная миотония).
Существует несколько форм, миотоний, при которых помимо миотонических симптомов, имеются признаки поражения других органов или систем.
В настоящее время показано, что большинство миотоний относятся к группе болезней, обусловленных нарушением функционирования ионных каналов (натриевых, калиевых, кальциевых и хлорных) в мембране скелетных мышц. Часть нозологических форм наследственных миотоний являются аллельными вариантами гиперкалиемического и гипокалиемического периодических параличей. К болезням хлорных каналов относятся миотонии Томсена и Беккера, миотоническая дистрофия. Типичным примером болезни натриевых каналов служат флюктуирующая миотония, ацетазоламидчувствительная миотония и группа парамиотоний.
ВРОЖДЕННАЯ МИОТОНИЯ
Распространенность врожденной миотонии во всем мире составляет 1:100000. Наблюдается увеличение распространенности в Северной Финляндии (7,3:100000).
Заболевание встречается в двух вариантах – миотония Томсена с аутосомно-доминантным типом наследования и миотония Беккера с аутосомно-рецессивным типом наследования. Показано, что эти заболевания являются аллельными вариантами, а эффект доминантности и рецессивности обусловлен различными мутациями в одном и том же гене.
1 вариант описал у членов своей семьи в 1876 г. датский врач Thomsen J. На основании генеалогического анализа у 64 больных в 7 поколениях он убедительно показал аутосомно-доминантный тип наследования болезни.
В 1966 г. Becker P. описал семью со сходными клиническими проявлениями и аутосомно-рецессивным типом наследования.
Ген заболевания CLСN1 расположен на хромосоме 7q35, содержит 23 экзона и кодирует белок хлорных каналов.
Белок мышечных хлорных каналов регулирует электрическую возбудимость скелетных мышц. При снижении или отсутствии функциональной активности белка нарушается проникновение хлора в мышечное волокно. Без аниона хлора, увеличивается проводимость и проницаемость для катионов. Катион калия накапливается в Т-трубочках, что приводит к повышенной деполяризацию мембраны. Деполяризация может достичь пороговой и привести к спонтанному срабатыванию потенциалов действия и повышению контрактильности мышцы.
ВРОЖДЕННАЯ АУТОСОМНО-ДОМИНАНТНАЯ МИОТОНИЯ ТОМСЕНА
1 вариант – врожденная миотония Томсена. Первые симптомы заболевания можно отметить с рождения или в периоде новорожденности.
Основным проявлением заболевания является миотонический спазм в различных группах мышц, возникающий после их интенсивного произвольного сокращения. Пассивные и произвольные движения, совершаемые с небольшим усилием или медленно, миотоническим спазмом не сопровождаются.
Наиболее часто первые признаки заболевания возникают в дистальных отделах рук. По мере прогрессирования заболевания в патологический процесс вовлекаются мышцы ног, туловища, шеи, мышцы языка, а также глазодвигательная, жевательная и мимическая мускулатура. Состояние тонического спазма измеряется в большинстве случаев секундами и почти никогда не длится свыше одной-двух минут.
Как правило, начальные симптомы миотонии родители замечают у ребенка в младенческом возрасте: 1) первые сосательные движения совершаются медленно, а затем становятся более энергичными; 2) после падения во время игры ребенок долго не может встать; 3) чтобы взять у ребенка какой-либо захваченный им предмет, необходимо насильно разжимать ему пальцы.
При ходьбе по ровному полу затруднения возникают особенно часто после длительного покоя, а также при смене темпа или характера движения. Миотонические спазмы в руках затрудняют письмо, рукопожатие и другие манипуляции. В школе ребенок не может сразу начать писать или быстро встать из-за парты. Эти дети не участвуют в играх, требующих быстрых движений.
Ценным тестом для выявления миотонии является ходьба по лестнице. Подняться на первую ступеньку для больного представляет большую трудность, чем дальнейший подъем по лестнице: больной «замирает», это время, которое необходимо для расслабления мышц.
Миотонический феномен может проявиться в артикуляции при произношении первых нескольких слов (смазанность речи); во время глотания (первые глотательные движения). Иногда затрудняются первые жевательные движения и т.д. У некоторых больных миотонический спазм наблюдается в круговых мышцах глаз (рис. 1). Попытка открыть глаза после крепкого зажмуривания затруднена; возникающая миотония ликвидируется постепенно, иногда через 30–40 сек. В ряде случаев, особенно при попытке произвести быстрое движение у больных возникает генерализованный миотонический спазм, во время которого может произойти резкое падение больного, сопровождающееся общей скованностью.
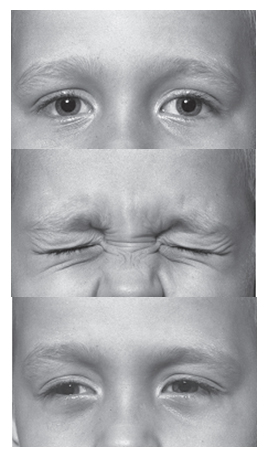
Рисунок 1. Миотонический спазм круговой мышцы глаз.
Выраженность спазма наибольшая в начале движения и уменьшается во время повторных мышечных сокращений. Усиление миотонического феномена возникает на холоде, уменьшение – в тепле, во время отдыха и при приеме небольших доз алкоголя.
Мышечная система больных обычно гипертрофирована, пациенты имеют вид атлетически сложенных людей. При этом мышечная сила может быть снижена. Часто больные жалуются на боли в различных группах мышц. Довольно типичны крампи в икроножных мышцах.
Характерным проявлением миотонии является повышение механической и электрической возбудимости мышц. Клинически это проявляется миотоническим феноменом, «перкуссионной миотонией» и «миотоническими разрядами» при ЭМГ-исследовании.
Перкуссионная миотония выявляется при ударе неврологическим молоточком. На месте удара образуется характерный мышечный валик, заметный в дельтовидных, ягодичных мышцах, мышцах бедра, голени и, особенно, в мышце языка. Рядом с валиком возникает мышечное углубление. Длительность миотонической «ямки» отражает тяжесть миотонии в целом.
Наиболее простой феномен – приведение большого пальца при ударе молоточком по тенару.
Наглядным также является тест с резким сжатием кисти в кулак, после которого больной не может сразу разжать пальцы. Замечено, что после сна миотонические симптомы могут временно усилиться.
Dupre с соавт. (2009) сообщил о 9 пациентах из 4 неродственных семей с аутосомно-доминантной миотонией, вызванных гетерозиготной мутаций хлорного канала 1 (CLСN1). Средний возраст начала болезни составил 13 лет (диапазон от 2 до 20). Наиболее распространенные клинические особенности включали перкуссионную миотонию (44%), миотонию при сжатии кисти (56%), «феномен разминки» (100%), генерализованную гипертрофию (78%), генерализованную мышечную скованность (78%) и обострение на фоне низких температур (56%). Менее часто наблюдались миотония при открытии глаз (11%), миотония век (22%), миотония языка (22%) и мышечные боли (11%). Никто из больных не жаловался на слабость, и никто не использовал лекарства, чтобы облегчить эти симптомы. Около половины пострадавших женщин отметили обострение симптомов во время менструации или беременности, и облегчения симптомов после менопаузы.
Течение миотонии Томсена обычно доброкачественное. В ряде случаев отмечается слабое прогрессирование заболевания.
Myotonia levior – «легкая миотония», мягкая форма аутосомно-доминантной миотонии, была впервые описана de Jong (1966). У больных отмечали миотонию век, дистальных отделов рук, без мышечной слабости, гипо- или гипертрофий. ЭМГ показывала миотонический феномен. Биопсии мышц, КТ бедра и мышц ног были нормальными.
Предполагается, что Myotonia levior – это вариант «болезни Томсена» с низкой экспрессивностью.
ВРОЖДЕННАЯ АУТОСОМНО-РЕЦЕССИВНАЯ МИОТОНИЯ БЕККЕРА
Второй вариант – миотония Беккера. Болезнь Беккера является более распространенной и более тяжелой, чем болезнь Томсена. В большинстве случаев заболевание возникает в возрасте от 4 до 13 лет. Первые признаки миотонии возникают в мышцах ног, через несколько лет – в мышцах рук. Отмечено вовлечение в процесс лицевой мускулатуры и дисфагия. Клинические проявления заболевания сходны с таковыми при миотонии Томсена, однако более выражены и у части больных сопровождаются миалгией. Мышечные гипертрофии встречаются в мышцах нижних конечностей.
Клинические проявления врожденной миотонии Томсена и Беккера представлены в таблице 1.
Таблица 1. Наследование и клинические проявления врожденной миотонии
|
Категория |
Клинические проявления |
|
|
Врожденная миотония |
Миотония Томсена Высокая изменчивость фенотипа и тяжести |
Миотония Беккера Высокая изменчивость фенотипа и тяжести |
|
Наследование |
Аутосомно-доминантное |
Аутосомно-рецессивное |
|
Дебют |
Начало в детстве и молодом возрасте |
Начало в детстве |
|
Краниальные симптомы |
Миотония век |
Миотония век |
|
Миотония языка |
Миотония языка |
|
|
Желудочно-кишечный тракт |
– |
Дисфагия |
|
Миотонический феномен |
Миотония (как правило, возникает во время быстрых произвольных движений мышц после периода покоя) Скованность и спазм мышц Задержка релаксация мышечных волокон после сокращения Миотония наиболее ярко проявляется в конечностях Миотония уменьшается при продолжающейся активности («феномен разминки”) Перкуссионная миотония Миотония сжатия (феномены «рукопожатия» и «зажмуривания») |
|
|
Мышечная слабость |
Нет мышечной слабости |
Транзиторная мышечная слабость, особенно в кистях и руках у 75% пациентов |
|
Миалгии |
Мышечные боли (реже) |
Мышечные боли характерны |
|
Гипертрофии мышц |
Мышечная гипертрофия («атлетическое» телосложение) |
Гипертрофия мышц нижних конечностей |
|
ЭМГ |
ЭМГ выявляет спонтанную, повторяющуюся электрическую активность («миотонический разряд») |
|
|
Усугубляющиефакторы |
Холодная температура усугубляет симптомы У женщин обострение симптомов во время менструального периода и беременности |
|
|
Облегчающие факторы |
Теплая погода и алкоголь являются факторами, уменьшающими симптомы У женщин уменьшение симптомов после менопаузы |
|
|
Степень тяжести |
Аутосомно-доминантный тип менее распространен и является менее тяжелым |
Аутосомно-рецессивный тип более распространен и является более тяжелым |
|
Молекулярный базис |
Мутация гена хлорного канала 1 скелетной мышцы (CLCN1) на хромосоме 7q35 |
|
В отличие от болезни Томсена, пациенты с болезнью Беккера имеют преходящую проксимальную мышечную слабость. Слабость возникает во время первой попытки определенного движения после периода бездействия и сохраняется в течение нескольких секунд. Исследование показало, что преходящая слабость представляет собой блок деполяризации, связанный с длительным восстановлением возбудимости мышечной мембраны.
У части больных может быть незначительное увеличение уровня активности креатинкиназы в плазме крови.
Электрофизиологические данные при врожденной миотонии: ЭМГ выявляет специфический миотонический феномен. В ответ на механическую или электрическую стимуляцию возникает повышенная возбудимость и повторяющиеся потенциалы действия в отдельных мышечных волокнах. Проведение игольчатой ЭМГ демонстрирует один из самых характерных для миотонии феноменов – миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика», которые возникают при введении и перемещении игольчатого электрода.
На ЭМГ продолжительность миотонического разряда при врожденной миотонии регистрируется от менее 1 сек. и до 10 сек. в отличие от миотонической дистрофии, при которой продолжительность миотонического разряда – более 2 сек. – до 30 сек.
Специфические морфологические признаки отсутствуют. В большинстве случаев выявляется вариабельный диаметр мышечных волокон, их гипертрофия и централизация ядер.
Лечение врожденной миотонии.
Целью лечения миотонии является снижение выраженности миотонических проявлений. Многие пациенты с недистрофической миотонией имеют слабые симптомы, которые не требуют лечения.
Необходимо также исключить провоцирующие факторы и препараты, которые усиливают миотонический феномен, такие как переохлаждение, β2-aгонисты, монокарбоновые аминокислоты, деполяризующие миорелаксанты.
Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа.
Если миотонические проявления, боль и слабость являются достаточно серьезными, могут использоваться противоэпилептические, антиаритмические препараты и блокаторы натриевых каналов.
Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений в симптоматическом лечении миотонии были использованы мексилетин, ацетазоламид, фенитоин, карбамазепин.
Проведенные наблюдения и рандомизированные исследования показали, что мексилетин и токаинид были самым мощными противомиотоническими средствами. Однако использование токаинида было ограничено и запрещено в связи с риском токсического воздействия на костный мозг и интерстициального заболевания легких.
Мексилетин используется наиболее часто. Он может быть полезным для предотвращения болевых феноменов и переходящей слабости, которая сопровождает миотонию Беккера. Начальная доза мексилетина 150 мг в день и более, с максимумом 300 мг три раза в день. Основные побочные эффекты: головокружение, диарея и диспепсия.
До начала приема мексилетина для оценки нарушений проводимости и интервала QT должна быть проведена ЭКГ.
Мексилетин противопоказан в случае второй или третьей степени атриовентрикулярной блокады. Его следует использовать с осторожностью у пациентов с первой степенью атриовентрикулярной блокады, дисфункцией синусового узла, или внутрижелудочковых дефектов проводимости.
Во время лечения должны проводиться анализы крови, функциональные исследования печени и ЭКГ в динамике.
Ацетазоламид назначают в дозе 125 мг / день и более, в случае необходимости до 750 мг (по 250 мг три раза в день).
Положительный эффект был получен при использовании ботулинического токсина у больных со спазмом круговой мышцы глаз.
В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенном введении человеческого иммуноглобулина (400 мг/кг) и преднизолона (1 мг/кг/сутки).
МИОТОНИЧЕСКАЯ ДИСТРОФИЯ
Миотонические дистрофии (МД) представляют собой группу наследственных мультисистемных заболеваний (глаз, сердца, головного мозга, эндокринной, желудочно-кишечного тракта, матки, кожи), которые разделяют по основным нарушениям функций: миотонии, слабости и раннем развитии катаракты (моложе 50 лет) (табл. 2).
В 2000 году Международный консорциум миотонической дистрофии (IDMC, 2000) разработал новую номенклатуру и ведущие принципы анализа ДНК.
Классическая форма миотонической дистрофии тип 1 (Steinert) встречается наиболее часто (98%) и является результатом увеличения нестабильных тринуклеотидных повторов CTG (до нескольких тысяч) в нетранслируемой области DMPK (Myotonin protein kinase). Ген заболевания МД1 картирован на 19 хромосоме.
Пациенты с клинической картиной миотонической дистрофии тип 2 / проксимальной миотонической миопатии (ПРММ) / синдром Ricker, которые имеют положительный тест ДНК с увеличением нестабильности четырех нуклеотидных повторов CCTG на 3 хромосоме, в настоящее время классифицируются как имеющие МД2/ПРММ.
Клинические черты типов миотонической дистрофии, связанной с нестабильностью увеличения нуклеотидных повторов, представлены в таблице 2.
Миотоническая дистрофия тип 1
Заболевание впервые описано Россолимо в 1901 г. В 1909 г Steinert H. и Battеn F. независимо друг от друга представили полное описание клинических проявлений. В различных популяциях заболевание встречается с частотой от 1 до 8 случаев на 40 тысяч населения. Тип наследования аутосомно-доминантный. Ген заболевания картирован на хромосоме 19q13.2-q13.3, содержит 15 экзонов и обозначается как DMPK ген. Единственный идентифицированный тип мутаций – экспансия тринуклеотидных СТG повторов в нетранслируемой области гена. В норме количество таких повторов колеблется от 5 до 37. У больных количество повторов находится в интервале от 50 до 2000. Отмечена корреляция между количеством СТG повторов и тяжестью течения заболевания. Так при мягких формах болезни количество повторов – от 50 до 200, при формах с поздним началом – от 300 до 1000, при врожденных вариантах болезни – от 1500 до 2000.
Экспрессируемый геном белок миотонин-протеинкиназа относится к семейству серин/треонин протеинкиназ и состоит из 624 аминокислот. Считается, что этот белок играет важную роль в регуляции клеточной дифференцировки и репликации ДНК.
Указанный повтор характеризуется значительной нестабильностью, наиболее выраженной в женском мейозе. Особенностью наследования заболевания является геномный импринтинг, характеризующийся различиями в тяжести течения заболевания в зависимости от того, от кого из родителей унаследовано заболевание. При передаче мутантного гена через матерей отмечается более выраженное течение болезни и возникновение врожденных форм. Серин/треонин протеинкиназы, кодируемые геном DMPK, присутствуют не только в скелетных мышцах, но и в миокарде, а также ЦНС. Этим и объясняются основные клинические проявления МД1.
Заболевание встречается в двух клинических формах: врожденной и классической.
Врожденная форма МД1. Первые признаки заболевания возникают еще во внутриутробном периоде и характеризуются резким снижением двигательной активности плода. Во время беременности у женщин отмечают многоводие, повышенную частоту предлежания плаценты и выкидышей.
Врожденная форма проявляется при рождении с дыхательной недостаточностью, нарушением сосания, диффузной мышечной гипотонией, косолапостью, повышенным риском внутримозгового кровоизлияния и эвентрацией диафрагмы. Наблюдается поражение жевательной, мимической и глазодвигательной мускулатуры и мышц дистальных отделов конечностей.
Характерным признаком является респираторный дистресс. В перинатальном периоде у младенцев с врожденной МД1 может потребоваться непрерывная искусственная вентиляция. Дети, которые остаются ИВЛ-зависимыми после 4-недельного возраста, имеют плохой прогноз для выживания.
В период новорожденности преобладающим в клинической картине является миопатический синдром, особенностью которого является сохранность или даже повышение сухожильных рефлексов. Признаки миотонии, как правило, присоединяются позднее. Во время первых 2 лет жизни дети с врожденной МД1 имеют трудности при вскармливании и подвергаются повышенному риску аспирационной пневмонии.
Характерна задержка темпов приобретения двигательных навыков. Заболевание достаточно быстро прогрессирует, часто приводя к внезапной смерти больных в раннем возрасте.
Среди больных с врожденной МД1 распространены нарушение слуха, умственная отсталость и неспособность к обучению. Определение степени когнитивной недостаточности требует тщательной оценки, потому что эти пациенты имеют выраженную слабость мышц лица, снижение слуха и не в состоянии хорошо говорить и общаться.
Классическая форма. Заболевание манифестирует в широком возрастном диапазоне – от 5 до 35 лет и характеризуется сочетанием симптомов миопатии, миотонии, сердечно-сосудистых, эндокринно-вегетативных нарушений и катаракты.
Миотонические признаки проявляются в виде миотонических спазмов и миотонических механических реакций. Миотонические спазмы возникают преимущественно в сгибателях пальцев кистей и жевательной мускулатуре. По мере течения заболевания симптомы миотонии уменьшаются и в поздних стадиях заболевания могут полностью исчезнуть.
Симптомы миопатии характеризуются слабостью и атрофиями различных мышечных групп. Наиболее часто процесс локализуется в мышцах лица, грудинно-ключично-сосцевидной мышце, надостных, подостных и височных мышцах. В ряде случаев в процесс вовлекаются глазодвигательные мышцы. Кожные покровы обычно имеют сероватый оттенок. Лицо больного приобретает маскообразное печальное выражение. По мере прогрессирования заболевания отмечается поражение бульбарных мышц, с возникновением дисфонии и затруднением глотания. Атрофические процессы в скелетной мускулатуре наиболее выражены в дистальных отделах конечностей.
Атрофические процессы и миотонические спазмы могут также возникать в дыхательной мускулатуре, что приводит к ограничению подвижности грудной клетки и снижению вентиляции легких. Это способствует гипоксии, возникновению аспирационной пневмонии, а также приступов апноэ во время сна.
У больных отмечается повышение частоты встречаемости дисморфических черт строения скелета, лицевого черепа, изменение дерматоглифики.
У 50% больных возникают сердечно-сосудистые нарушения, главным образом в виде гипертрофии желудочков, симптомов нарушения проводимости и сердечного ритма.
У значительного числа больных возникают поражения глаз в виде катаракты, блефарита, конъюнктивита, помутнения роговицы.
Эндокринные нарушения характеризуются гипогонадизмом, азооспермией и снижением либидо у мужчин, и нарушением менструального цикла, гирсутизмом и ранним климаксом у женщин. Характерным симптомом является облысение, сопровождающееся изменением структуры волос. У мужчин наибольшее выпадение волос отмечается в области лба и висков, для женщин характерно гнездное или диффузное облысение.
У больных отмечается снижение интеллекта, психиатрические и поведенческие нарушения в виде обсессивно-компульсивных и пассивно-агрессивных черт личности. У 50–90% пациентов уровень IQ равен 40–80.
Желудочные гипокинезии с хроническими запорами, преходящие симптомы со стороны мочевых путей, такие как неудержание мочи или частое мочеиспускание, являются общими при МД1.
Пациенты с МД1 обладают повышенной чувствительностью к седативным средствам, особенно барбитуратам и опиатам.
На ЭМГ выявляются специфическая миотоническая задержка (продолжительность миотонического разряда – более 2 сек. до 30 сек.), снижение количества функционирующих двигательных единиц и скорости проведения возбуждения по эфферентным волокнам периферических нервов.
Таблица 2. Дифференциальная диагностика типов миотонической дистрофии, связанной с нестабильностью увеличения числа нуклеотидных повторов
|
Заболевание |
Миотоническая дистрофия тип 1 (Steinert) |
Миотоническая дистрофия тип 2/ ПРММ (Ricker) |
|
Эпидемиология |
Широко распространена |
Европа |
|
Наследование |
Доминантное |
Доминантное |
|
Генный дефект |
Хромосома 19q13.3; Мутантный ген DMPK Тип мутации – увеличение CTG; Частота повторов колеблется от 50 до 4000; Норма 5–37 повторов |
Хромосома 3q21; Мутантный ген ZNF9 Тип мутации – увеличение CCTG Частота повторов – от 78 до> 11000; Норма <75 повторов |
|
Антиципация |
Выраженная |
Мягкая |
|
Дебют |
Широкий диапазон (от младенчества до взрослого возраста) Начало наиболее тяжелых случаев с младенчества |
Широкий диапазон возрастов (от 8 до 60 лет) |
|
Врожденные формы |
Часто |
Редко |
|
МЫШЦЫ: Слабость мышц |
Генерализованная слабость, гипотония, респираторный дистресс у младенцев |
Мягкая |
|
Лицо |
12% (легкая) |
|
|
Птоз |
Легкий |
|
|
Грудинно-ключично-сосцевидная |
Вариабельно |
|
|
Проксимальные отделы |
В поздних стадиях |
Редко |
|
Дистальные отделы |
Кисти |
|
|
Другая локализация |
||
|
Гипертрофии мышц |
Отсутствуют |
Икроножные мышцы |
|
Мышечная боль и дискомфорт |
Вариабельно |
56% (боль возникает и без миотонии, провоцируется холодом, пальпацией) |
|
Миотония |
В первую очередь кисти, мышцы предплечья и язык; иногда дыхательные мышцы и гладкая мускулатура кишечника или матки. Миотонию уменьшает тепло и повторные упражнения |
В основном в кистях и бедрах; вариабельна; часто трудно обнаружить. Миотония уменьшается при повторных сокращениях |
|
Провокационный стимулы |
Миотония усугубляется отдыхом и холодом; тяжесть миотонии относительно постоянна в пострадавших мышцах |
Миотония усугубляется отдыхом, иногда отсутствует при клиническом исследовании; наиболее постоянны тест рукопожатия и миотония бедер |
|
Мультисистемные признаки: Катаракта |
(100%) |
|
|
Облысение |
||
|
Нарушение слуха |
/- |
20% |
|
Кардиальные аритмии |
Вариабельно (20%) |
|
|
Желудочно-кишечный тракт |
Холелитиаз, периодическая кишечная псевдообструкция, нарушение вскармливания при врожденной форме |
Дисфагия |
|
Гипогонадизм |
20% |
|
|
Гиперсомния |
Вариабельно |
|
|
Гипергидроз |
Вариабельно |
|
|
Когнитивные, психиатрические и поведенческие нарушения |
Мягкие и тяжелые От 50 до 90% Обсессивно-компульсивные и пассивно-агрессивные черты личности |
Мягкие или отсутствуют |
|
Гипергликемия |
20% |
|
|
Биопсия мышц: вакуолизированные ядра |
Разнообразные мышечные волокна, но преобладает 1 тип |
2 тип мышечных волокон |
|
ЭМГ: миотония |
||
|
МРТ – ЦНС |
Белое и серое вещество |
Белое вещество |
|
Симптоматическая терапия |
Общеукрепляющая; ортезы; удаление катаракты; мониторинг аритмии и дыхательной недостаточности; кардиостимулятор; антимиотоническая терапия (мексилетин) |
Удаление катаракты; иногда потребность в кардиостимуляторе; антимиотоническая терапия часто не нужна, но при боли и спазме (карбамазепин или мексилетин, нестероидные противовоспалительные) |
|
Хирургическое вмешательство |
Избегать деполяризующих миорелаксантов, опиатов, и барбитуратов при хирургических вмешательствах; тщательно контролировать во время и после операции возникновения спазма, аритмии и апноэ; возможность развития острого некроза скелетных мышц |
|
Типичным морфологическим дефектом, выявляемым даже в начале заболевания, являются выраженное уменьшение размеров мышечных волокон 1 типа и значительное увеличение размеров волокон 2 типа. В центре мышечных волокон определяются саркоплазматические включения, имеющие вакуолизированные ядра. Под сарколеммой мышечного волокна обнаруживаются специфические агрегаты тубул в виде пчелиных сот.
МРТ: при врожденной форме МД1 – увеличение размеров желудочков с рождения. У некоторых больных гипоплазия мозолистого тела. Вовлечение лимбической системы. Уменьшение серого вещества в лобной и теменной долях, таламусе и гиппокампе. Глобальная церебральная атрофия.
Частота поражения белого вещества при МД более 50%. Степень коррелирует с выраженностью психомоторных нарушений, возрастом и длительностью заболевания. Более тяжелые изменения со стороны серого и белого вещества головного мозга наблюдаются в случае МД1, чем при МД2.
Возможна дородовая диагностика (анализ ДНК, выделенной из амниотической жидкости или хориона) и определение носительства мутации на доклинической стадии у родственников больного.
Лечение
Лечение пациентов с врожденной МД включает респираторную поддержку и использование пищевого зонда.
Коррекция косолапости осуществляется с применением ортезов и/или корректирующей операции деформированной стопы.
Необходим контроль степени когнитивного дефицита и нарушения слуха.
Проблемы с речью и боль в животе купирует прием антимиотонических средств. Мексилетин используется в борьбе с некоторыми из симптомов, особенно в позднем детстве или в подростковом возрасте, когда у пациентов может быть спазм в жевательных мышцах, рецидивирующий вывих нижней челюсти и боль в мышцах. Для выявления скрытой аритмии в период лечения мексилетином пациентам необходимо проводить электрокардиографический контроль.
После использования общей анестезии существует повышенный риск сердечной аритмии и апноэ, что требует суточного мониторинга. Апноэ может развиваться через несколько часов после того, как пациент был экстубирован.
Ограничение физической активности в детстве не является необходимой мерой, при условии, что ребенок имеет достаточную устойчивость и координацию.
Миотоническая дистрофия тип 2 / Проксимальная миотоническая миопатия (синдром Ricker)
МД2 является результатом нестабильного увеличения повторов четырех нуклеотидов – CCTG, гена ZNF9 на хромосоме 3q21.
Число CCTG-повторов обычно в ДНК лейкоцитов менее 75. Число у больных с МД2 составляет от 78 до более чем 11 000 повторов. Молекулярные механизмы, ведущие к проявлениям МД2, как полагают, аналогичны МД1, и относятся к токсическому эффекту аномально расширенной РНК, которая накапливается в ядрах мышечных клеток.
МД 2/ПРММ обычно дебютирует во взрослом возрасте и имеет вариабельные проявления, такие как раннее начало катаракты (моложе 50 лет); миотонию, напряжение мышц бедер, боли в мышцах, а также слабость (сгибателей и разгибателей бедра, мышц живота или длинных сгибателей пальцев) различной степени выраженности.
Симптомы часто появляются между 20 и 70 годами, и пациенты, связывают их с чрезмерной нагрузкой на мышцы, “радикулитом”, артритом, фибромиалгией или приемом статинов. Боль возникает без очевидной причины, как правило, колеблется по интенсивности и локализации в конечностях. Болевые проявления могут длиться от нескольких дней до нескольких недель.
В дебюте МД2 имеет место только легкая слабость мышц лица, сгибателей и разгибателей мышц бедра и сгибателей пальцев. Часто фиксируют слабость сгибателей шеи. Гипотрофия лицевых мышц и мышц конечностей незначительная. Иногда отмечается гипертрофия икроножных мышц. По мере прогрессирования заболевания возникает проблема с приседанием.
Миотонический феномен при МД2 часто менее очевиден, по сравнению с пациентами с МД1. Миотония варьирует от минимальной до умеренной. Тяжесть прогрессирует от нескольких дней до нескольких недель. Прямая перкуссия мышц предплечья и тенара является наиболее чувствительным клиническим тестом для выявления миотонии, но может и отсутствовать. Миотония сцепления проявляется не постоянно. В случаях с поздним началом МД2 миотонию можно выявить только на электромиографии в результате тестирования нескольких мышц.
Другие проявления, такие как повышенная потливость, гипогонадизм, нарушение толерантности к глюкозе, дисфагия, нарушения сердечной проводимости, когнитивные и нейропсихологические нарушения также могут возникнуть со временем и ухудшить течение заболевания.
Среди больных женщин сообщается о более высокой частоте преждевременных родов и недоношенных.
В настоящее время нет четких доказательств врожденной формы МД2. Тем не менее, в исследованиях родительско-детских пар есть свидетельства МД2 с более ранним началом известных симптомов. Клинические проявления возникают в детстве. Они, как правило, транзиторные и характеризуются миотонией в руках и бедрах. Проксимальная слабость, мышечные боли, катаракта, сердечная аритмия обычно не развиваются до середины взрослой жизни или старости.
Когнитивные трудности также возникают при МД2, как при МД1, но проявляются во взрослом возрасте и связаны со снижением мозгового кровотока во фронтальной и передней височной долях, и уменьшением объема мозга.
Клинические лабораторные тесты .
Для диагностики МД2 используется ДНК-тестирование.
Обследование с помощью щелевой лампы проводится для раннего выявления формирующейся катаракты.
Электромиография: показательно исследование мышц шеи, большеберцовой мышцы и группы передних мышц бедра.
У некоторых пациентов имеет место увеличение (в 2-8 раз) уровней креатинкиназы, гамма-глютамилтрансферазы, холестерина, AЛT и лактатдегидрогеназы. Снижен креатин и общий белок.
Пациенты с повышенным уровнем креатинкиназы могут иметь больше послеоперационных осложнений.
Биопсия мышц при МД2 в сравнении с классической МД1 указывает, что атрофия менее тяжелая. По-видимому, при МД2 предрасположенность к атрофии имеют 2 тип мышечных волокон, а при МД1 атрофируются волокна 1 типа.
МРТ: Белое вещество гиперинтенсивно на Т2 взвешенных изображениях. Уменьшение серого вещества в лобной и теменной долях, аномальное серое вещество таламуса и гиппокампа. Степень лейкопатии и атрофии коррелирует с выраженностью когнитивного и моторного дефицита, но менее тяжелое, чем при МД1.
Лечение
В целом, наблюдение при МД2 является похожим на МД1, но существует меньше потребности в поддерживающей терапии, такой как общеукрепляющие препараты, ортезы и средства передвижения.
Требуется динамическое наблюдение окулистом и серийный ЭКГ-контроль, мониторинг гипогонадизма и инсулинорезистентности.
Миотония при МД2 менее выражена, но при определенных обстоятельствах, используется противомиотоническая терапия, особенно если беспокоит боль и постоянно присутствует или часто возникает спазм мышц. Карбамазепин или мексилетин, наряду с нестероидными противовоспалительными средствами купируют эти проявления.
Список литературы:
1. Вельтищев Ю.Е., Темин П.А. Наследственные болезни нервной системы// Руководство для врачей. – Москва, “Медицина”. – 1998. – 497 с.
2. Феничел Дж.М. Педиатрическая неврология: Основы клинической диагностики: Пер. с анг.- М.: ОАО «Медицина», 2004.
3. Day, J. W., Ricker, K., Jacobsen, J. F., Rasmussen, L. J., Dick, K. A., Kress, W., Schneider, C., Koch, M. C., Beilman, G. J., Harrison, A. R., Dalton, J. C., Ranum, L. P. W. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum.// Neurology.-2003.-Vol.60.-Р.657-664.
4. Donahue, L. A., Mangla, R., Westesson, P.-L. Neuroimaging in myotonic dystrophy type 1.//Neurology. – 2009. – Vol.73. – P.1931.
5. Dupre, N., Chrestian, N., Bouchard, J.-P., Rossignol, E., Brunet, D., Sternberg, D., Brias, B., Mathieu, J., Puymirat, J. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. //Neuromusc. Disord. – 2009. – Vol.19. – P. 330-334.
6. George, A. L., Jr., Crackower, M. A., Abdalla, J. A., Hudson, A. J., Ebers, G. C. Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita). //Nature Genet. – 1993. – Vol. 3. – P.305-310.
7. Lehmann-Horn, F., Mailander, V., Heine, R., George, A. L. Myotonia levior is a chloride channel disorder. //Hum. Molec. Genet. – 1995. – Vol.4.- P. 1397-1402.
8. Logigian, E. L., Martens, W. B., Moxley, R. T., IV, McDermott, M. P., Dilek, N., Wiegner, A. W., Pearson, A. T., Barbieri, C. A., Annis, C. L., Thorton, C. A., Moxley, R. T., III. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. //Neuroogy. – 2010. – Vol.74. – P.1441-1448.
9. Modoni, A., Silvestri, G., Pomponi, M. G., Mangiola, F., Tonali, P. A., Marra, C. Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1.//Arch. Neurol. – 2004. – Vol.61. P.1943-1947.
10. Tramonte, J. J., Burns, T. M. Myotonic dystrophy. //Arch. Neurol. – 2005. –Vol.62. – P.1316-1319.
11. Udd, B., Meola, G., Krahe, R., Thornton, C., Ranum, L., Day, J., Bassez, G., Ricker, K. Report of the 115th ENMC workshop: DM2/PROMM and other myotonic dystrophies. 3rd Workshop, 14-16 February 2003, Naarden, The Netherlands. //Neuromusc. Disord. – 2003. – Vol.13. – P.589-596.
12. http://www.ncbi.nlm.nih.gov/omim
13. http://neuromuscular.wustl.edu/